7月10日,国家药品监督管理局发布了《接受药品境外临床试验数据的技术指导原则》(下称《原则》)。这意味着,国家食药监总局药审中心在2017年10月20日发布的《接受境外临床试验数据的技术要求》征求意见稿采纳了合理意见建议后的正式落地。
去年年底,即征求意见稿出台不久,中国医学科学院药物研究所新药开发研究室主任、国家食品药品监督管理总局新药审评委员吴松在接受《人民日报》采访时表示,“共享”临床数据将极大减少进口新药在中国获批上市的时间,同时也有利于中国医药企业走向海外,开展境外临床试验。
接受境外临床数据到底利好哪些企业呢?笔者在与行业人士交流后发现,大家普遍认为利好的企业至少包括以下5类:(1)跨国大药企;(2)从海外引进在研项目的创新药企;(3)率先在美国、澳大利亚等境外开展临床试验的创新药企;(4)在印度等境外开展仿制药BE试验的企业;(5)开发危重疾病、罕见病、儿科的企业。
跨国大药企
过去,一些新药在我国的上市时间,平均要比欧美晚5到7年。当时部分原因是,中国并不认可在境外多中心取得的临床试验数据,国外新药首次进入中国,必须在国外做完Ⅱ期临床试验后才能在中国进行临床试验,在中国完成Ⅲ期临床试验后才能获批上市。
《原则》明确提出:只要是真实可靠、符合ICH GCP和药品注册检查要求;支持目标适应症的有效性和安全性评价;不存在影响有效性和安全性的种族敏感性因素的境外临床研究数据都能够被CDE完全接受。即使是存在影响有效性和/或安全性的种族敏感性因素,数据外推至中国人群的有效性和安全性评价存在较大的不确定性,CDE也可以部分接受。
这显然让“有条件”接受境外临床数据更明晰,从机制上缩短境外新药在国内上市的时间,毕竟数据可以在一定程度上通用了,患者或许也能以最快速度享受到优质的创新药品,而不用像以往一样等上数年或通过非正式渠道从境外获取“救命药”。
从海外引进在研项目的创新药企
近年来中国药企研发和资金实力逐渐增强,在国家鼓励创新的政策引导下,国内企业越来越多地与跨国企业建立合作,通过专利许可、共享权益等方式引入国际创新研发项目。
一方面,这种通过跨境合作引进的方式比自主研发更快捷,国内药企也是希望借助与海外企业合作的方式提升国际化研发能力。另一方面,跨国大药企有聚焦核心业务、剥离非核心研发项目资产的考虑,海外中小型生物医药公司则是希望与本土有研发实力的药企合作来快速拓展中国市场。
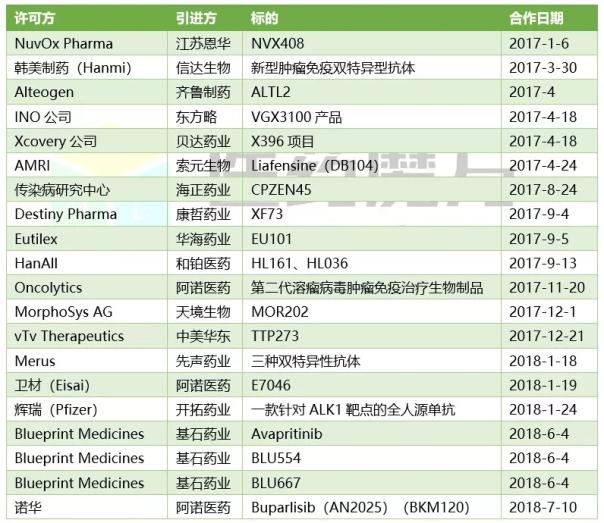
中国创新药海外引进案例(部分)
在刚刚过去的6月,基石药业一举从美国2家中型生物技术公司拿下商业化引进4个项目。该公司商务拓展负责人告诉记者,美国中小型生物技术并不具备在亚太地区商业化开发能力,他们通过与国内具有临床开发能力的创新药企合作,推动授权产品在大中华区的单药及联合治疗的临床开发和商业化。CDE认可境外数据,或将进一步加速产品在中国的上市进程。
在境外开展临床试验再回国内申报的企业
国内创新药开发企业在药品注册时一般采取两类策略:一是获得国家药监局临床批件后,在国内开展临床试验,结束后申报新药并在国内上市;二是同步开展中国和国外的临床研发,待试验结束后分别申请中国及国外新药批文,在全球同步上市。
一位新药开发负责人向记者透露:较高的研究标准和质量标准(特别是早期阶段的能力)、有可比性的成本、及时的审判批准和可靠的病人招募等因素使得澳大利亚成为极具吸引力的临床试验国家。即便是同步准备IND申报资料,在澳大利亚的推进速度可能会更快。
《原则》指出:鼓励开展境内外同步研发。境外临床试验数据可以用于支持需要进行有效性和/或安全性评价的各类注册申请。例如,在境外开展的早期临床研究数据,可用于支持在我国开展注册临床试验的申请。这对于创新药开发企业而言,显然是降低了开发成本。
鸿运华宁是一家十年如一日坚守开发原创性新药的企业。该公司新药开发负责人告诉记者,创新药在澳洲开展床新药的I期临床,具有快速、花费少、质量高的特点;申办方可降低研发成本,缩短研发周期,提高成功率。不过他也坦言,在健康受试者身上的试验优势,在后期进行患者试验时,会受到患者资源的限制。国家药监总局出台的新政,若能让创新药企借力于澳大利亚等国家的早期开发数据,支持后续在国内的注册申报,将会大大节省研发资源,避免重复浪费。
在境外开展仿制药BE试验的企业
在国家药品监督管理局发布的《接受药品境外临床试验数据的技术指导原则》解读中明确指出:境外完成的仿制药生物等效性(BE)试验数据,研究质量好,具备真实性、完整性、准确性和可溯源性的,也可用于在我国的注册申请。
据了解,在过去,国内制剂企业在出口美国、欧盟时,通过在印度等境外国家进行预BE/BE试验,不仅可以降低成本,还能提高数据被FDA/欧盟的认可度。一般这些“出海”产品被欧美批准后,再通过海外共线生产转报国内的“曲线回国”方式在国内上市。医药魔方记者为此也向行业内相关人士求证。
上海安必生制药董事长雷继峰在接受记者采访时表示:仿制药生物等效试验(BE)是比较两个处方在同一个人体(受试者)上的体内表现。BE试验结果与受试者人种种族无关,与处方工艺、BE试验设计有一定关系,欧美药监机构和WHO专家对此认识完全一致。欧美和WHO均接受世界任何国家符合GCP和GLP的BE数据。因此向FDA申报仿制药申请(ANDA),没有硬性规定必须在美国境内做BE,也可以在加拿大、印度等境外国家做,前提是只要符合GLP和GCP管理的临床试验基地,同时数据符合ICH要求。中国加入了ICH后,也同样要求在数据上与国际接轨。
曾经与印度CRO公司合作过100多项BE试验的雷继峰还透露,印度CRO产业高度发展,已形成规模化,大部分CRO公司能够像欧美公司一样,可以提供BE试验一站式服务。不过,由于印度受试者群体大,能加快入组速度;同时相比于欧美甚至中国,印度的BE服务费用更加低廉,是吸引越来越多药企到印度做BE的主要原因。据不完全统计,在向FDA申请ANDA的项目中,每年约有50%到60%是在印度做BE。
医药魔方记者获悉,早在两年前,泰格医药已经在印度成立子公司(见:泰格医药在印度成立子公司,提高临床试验数据管理、统计分析及医学写作能力)。一位仿制药开发的资深专业人士也向记者证实:除了FDA,还有欧盟都会认可在印度等境外国家的临床试验数据,只要数据真实性、完整性、准确性和可溯源性等符合ICH要求。如今,CDE直接认可印度的BE数据,显然降低了开发成本,也省去了企业“曲线回国”的路径,于国内因一致性评价“坐地涨价”的CRO企业而言,这一政策或带来一定打击。
雷继峰坦言,我国认可仿制药的境外BE数据对制药行业本身而言是好事,这不仅有利于国内药企降低临床开发成本;同时也能帮助老百姓早日吃上价格亲民的仿制药;尽管短期内对国外CRO有些镇痛,但长远看是有利于国内CRO学习国际规范,提高专业水平,并打破行业资源垄断。
开发危重疾病、罕见病、儿科的企业
《原则》在最后还特别提到,对于用于危重疾病、罕见病、儿科且缺乏有效治疗手段的药品注册申请,属于“部分接受”情形的,可有条件接受。
以开发“罕见病”治疗的孤儿药为例。此前上市的明星药物西达本胺是一款用于治疗复发或难治性外周T细胞淋巴瘤的罕见病药物。生产商微芯生物总裁兼首席科学官鲁先平曾在接受媒体采访时表示,由于公司与国家食药监总局药品审评中心沟通充分,西达本胺的审评审批速度得以加快。但即便如此,微芯生物也花了4年时间才完成了近100例病人的二期试验。到2015年西达本胺获准上市时,该药20年的专利独占期仅剩8年。
国家药监总局出台的新政提出对“部分接受”情形有条件接受,对那些海外临床数据“不完美”的孤儿药产品,有望在国内“有条件批准”上市。医药前不久也做了统计调研,越来越多的国内创新药企借力孤儿药政策,实现弯道超车。(见:这13个国产新药,获得了FDA孤儿药资格认定)。种种迹象表明,这显然是利好孤儿药开发企业的。
获得FDA孤儿药资格认定的国产创新药